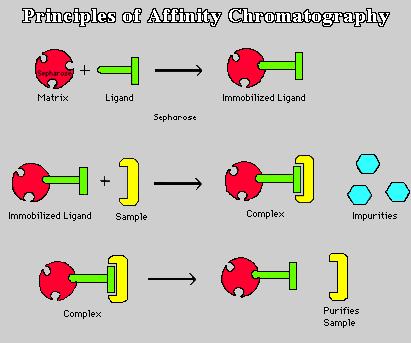
A biochip is a device that has some of the features of a computer chip but, instead of doing calculations, it uses living cells or molecules from living cells to greatly speed up certain laboratory tests. A typical biochip is a glass or plastic chip or tile a few inches on a side. It has hundreds or even tens of thousands of microscopic droplets of material stuck to its surface like gum on a sidewalk. A computer looks at the chip using a camera. Information from a biochip can be used to learn about the differences between genes, cells, or drugs. It can also be used to study many other questions about cells. Biochips are also called microarrays, where micro means ‘‘small’’ and an array is any regular grid, such as a chessboard. The droplets on a biochip are laid down in a checkerboard pattern. A square chip five inches on a side may have 40,000 or more spots on its surface.
A biochip is a device that has some of the features of a computer chip but, instead of doing calculations, it uses living cells or molecules from living cells to greatly speed up certain laboratory tests. A typical biochip is a glass or plastic chip or tile a few inches on a side. It has hundreds or even tens of thousands of microscopic droplets of material stuck to its surface like gum on a sidewalk. A computer looks at the chip using a camera. Information from a biochip can be used to learn about the differences between genes, cells, or drugs. It can also be used to study many other questions about cells. Biochips are also called microarrays, where micro means ‘‘small’’ and an array is any regular grid, such as a chessboard. The droplets on a biochip are laid down in a checkerboard pattern. A square chip five inches on a side may have 40,000 or more spots on its surface.The most common kind of biochip is the DNA microarray, also called a gene chip or DNA chip. Deoxyribonucleic acid (DNA) is the long, coded molecule used by all living things to pass on traits to offspring. DNA also tells each cell how to make all the molecules it needs to live, like a cookbook containing many recipes. The DNA of almost every living thing is at least slightly different from that of every other.
In one type of DNA chip, genes (short pieces of DNA that code for single molecules) are placed on the chip. Since even large molecules are too small to see with the naked eye, millions of copies of each gene can be placed on a tiny spot on the chip.
HOW DNA CHIP WORKS?
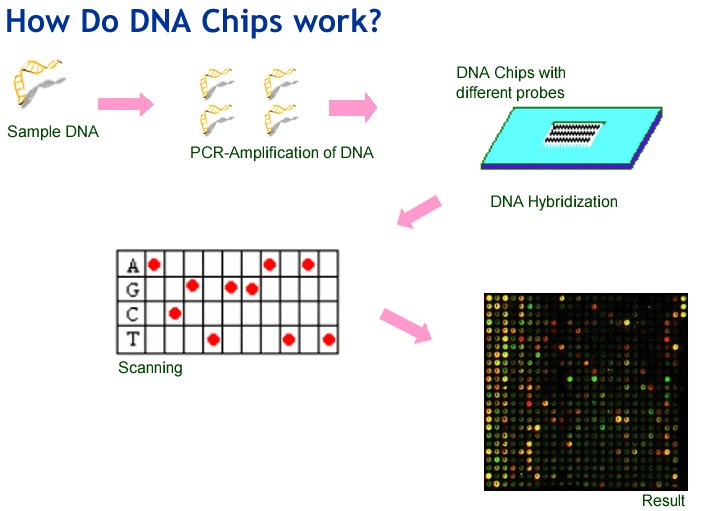
In a DNA chip, each separate spot (also called a probe) contains one type of defective gene. To find out if a person has any of these defective genes in their own DNA, DNA is taken from the person’s cells. Copies of the person’s DNA are made, and these copies are labeled, meaning that they include a chemical that glows when ultraviolet light (which is invisible to the eye) shines on it. Small drops of liquid containing labeled copies of the person’s DNA are then added to the spots on the biochip.
A normal DNA molecule is shaped like a ladder, but the DNA copies being mixed on the biochip are one-sided copies, like a ladder that has been sawed in half lengthwise, cutting every rung in half. When two pieces of one-sided DNA that have matching rungs (or bases, as they are called) meet, they lock or zip together. When this happens, the two pieces of DNA are said to hybridize. If the patient’s genes match any of the defective genes that have been put on the biochip, they will attach to (hybridize with) those defective genes.
The chip is then washed to remove any of the person’s DNA that has not found a match on the chip. Finally, the chip is placed in ultraviolet light, and a camera records any spots that glow. These are spots where the labeled copies of the person’s DNA have matched up with DNA on the chip.
Examining a patient’s DNA for defects is called genetic screening. By using a biochip, genetic screening can be done very quickly, all the tests can be done at once, rather than doing hundreds or even thousands of separate tests.
Genetic screening is only one way of using biochips. Another important use for biochips is to study how genes are used by living cells. Each gene tells the cell how to make a certain protein molecule. Cells read the recipe given by the gene by first making another molecule, mRNA, which copies the information in the gene. The mRNA can then go to a place in a cell that will build the molecule that the gene codes for. The more mRNA a cell has for a gene at a particular time, the more it is said to be ‘‘expressing’’ that gene, i.e, the more of that particular molecule it is making. Gene expression changes all the time for thousands of genes in every cell.
In the laboratory, scientists can make DNA molecules from the mRNA found in a cell. This matching DNA is called cDNA (complementary DNA). If a biochip has all the genes of an organism dotted on its surface, then cDNA made from the mRNA in a cell can attach to (hybridize with) the genes on the chip. The more a gene is being expressed in the cell, the more cDNA for that gene there will be, and the more that cDNA will stick to the matching genes on the biochip. Spots with more labeled cDNA will glow more brightly under ultraviolet light. In this way, scientists can literally take a snapshot of how the genes in a cell are being expressed at any one time—how much the cell is making, at that moment, of thousands of different substances. This is extremely useful in trying to understand how cancer cells grow and in many other medical problems.